Phylogenetic Tree Construction¶
Phylogenetic trees are useful tools in diversity analysis because they let us consider the evolutionary relationships between organisms. There are several ways in which trees can be constructed, and shown below are the common ways for constructing trees using 16S rRNA data.
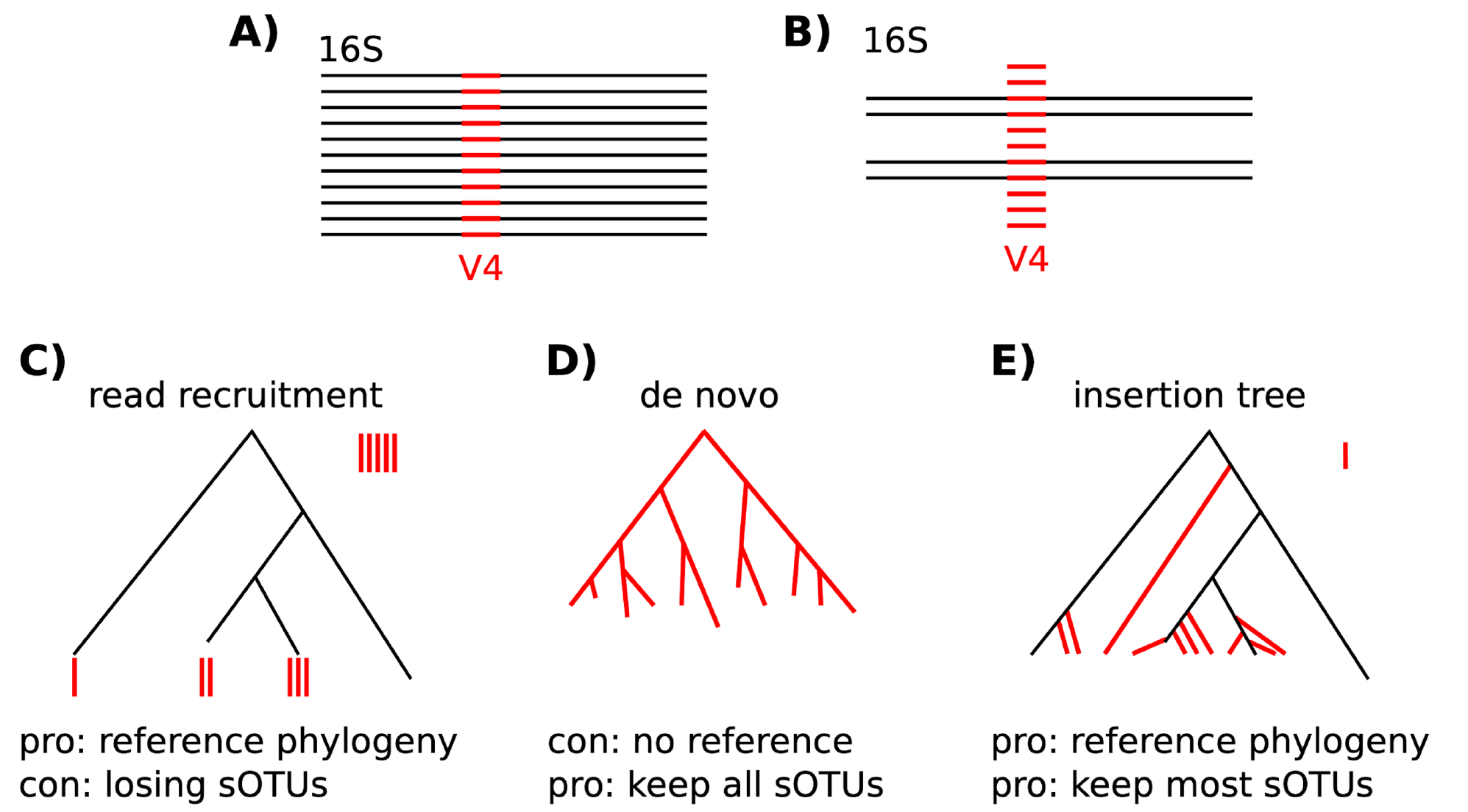
If you are only interested in a quick and dirty analysis, then you can use a de novo approach to create what is called a "fast tree" using the align-to-tree-mafft-fasttree pipeline in the phylogeny plugin in QIIME2. While we don't recommend this for publishing purposes, it can be useful for quickly exploring your data if needed because the insertion tree method can take a long time.
We will use the SEPP (SATe-enabled Phylogenetic Placement) method available through QIIME2’s fragment-insertion plugin to generate the rooted phylogenetic tree needed for phylogenetic diversity metrics such as Faith’s PD and UniFrac. SEPP places our ASVs into a backbone reference tree, in this case from Greengenes2, though the reference database can be changed with the --i-reference-database parameter. It attempts to match representative sequences from our samples to sequences already in the reference tree, and when an exact match is not available, it identifies the best branch point for insertion. This approach ensures that all sequences are incorporated into a tree with meaningful evolutionary structure, and it is recommended because it is generally more accurate and informative than building a de novo tree from our sequences alone.
Let's navigate to our tree directory and download the reference tree. You can look at your options at https://docs.qiime2.org/2021.11/data-resources/ under the SEPP reference databases heading at the bottom. As mentioned above, we are using the Greengenes2 database today, which can be found at https://ftp.microbio.me/greengenes_release/2022.10
Runtime
The SEPP placement step takes approximately 1.5 hours with 4 threads. During the workshop we copy a pre-built tree instead.
Get the SEPP Reference Database¶
First move into the tree directory
For a full description of all the options, plus some extras that we didn't use today, look at the documentation page.
Note that the plugin we are using is fragment-insertion, and the method is sepp. We use the representative sequences here, not the table, because this is completely independent of your samples; all it needs is a list of the sequences in your dataset and the ASV label associated.
As an output, we get the tree.qza that we need for diversity analyses. We also get a tree_placements.qza file, which has information about where it places your features before the actual rooted tree is generated. We get this because it is an intermediate file that is generated during the tree placement, but we don't actually need to look at it (unless you get really invested in your tree and phylogenetics in the future).
Run SEPP¶
This job can take a while (especially in real datasets), so we'll submit it as a job. Here, it may take around 20 minutes.
Move out of the tree directory and into the slurm directory
Now make a script for the tree job -nano is a command-line text editor and it allows you to create and edit files directly within your terminal.
This is what you will copy and paste into the sepp_script.sh
What do each of these lines mean?
#!/bin/bash
#SBATCH --job-name=sepp
#SBATCH --nodes=1
#SBATCH --ntasks=24
#SBATCH --partition=amilan
#SBATCH --time=12:00:00
#SBATCH --mail-type=ALL
#SBATCH --mail-user=$USER@colostate.edu
#SBATCH --output=slurm-%j.out
#SBATCH --qos=normal
#Activate qiime
module purge
module load qiime2/2026.1_amplicon
# go to your decomp directory
cd /scratch/alpine/$USER/decomp_tutorial
#frament insertion sepp
qiime fragment-insertion sepp \
--i-representative-sequences dada2/seqs.qza \
--i-reference-database tree/2022.10.backbone.sepp-reference.qza \
--o-tree tree/tree_gg2.qza \
--o-placements tree/tree_gg2_placements.qza \
--p-threads 4
to save this file after you copy the code into the file, use:
Then:
To submit the script use this command
Get the Pre-Built Tree (Workshop Shortcut)¶
For the workshop, copy the tree that was already built from this dataset:
Dataset Specificity
This pre-built tree was constructed specifically from the ASVs in this workshop dataset. Do not use it for your own data, you must run SEPP or another tree-building method on each new dataset.
Outputs¶
| File | Type | Description |
|---|---|---|
tree_gg2.qza |
Artifact | Rooted phylogenetic tree with ASVs placed |
tree_gg2_placements.qza |
Artifact | Detailed placement information (from SEPP) |
Next: Alpha Diversity